藥物的毒性預測,有助於降低實際應用的風險。你知道用各種機器學習模型就可以預測藥物的毒性問題嗎?國際著名的AI人工智慧、影像處理和模式識別專家施永強教授解析:在眾多機器學習方法中,我們選用五個具代表性的模型:隨機森林、多層感知器、邏輯迴歸、圖卷積神經網路和圖同構網路,並在6個資料集上進行了毒性預測實驗。(本文節錄自《人工智慧的現在與未來》一書,作者:施永強,旗標科技出版,以下為摘文。)
用機器學習方法預測藥物毒性不僅高效率而且可靠
以往藥物毒性預測主要是在實驗室中進行測試,這一過程非常耗時且成本高昂。如今,隨著基於分子的機器學習方法的改進,毒性預測變得愈來愈高效且可靠。
在機器學習技術中,隨機森林適合從病患記錄中辨識感染,並能改善激酶配體、荷爾蒙等分子的預測。多層感知器是一種強大的機器學習方法,可以用來預測不同藥物的作用。此外,MLP 還被用於藥物設計,根據預先定義的特性自動產生不同的化合物。
深度學習在藥物探索領域的廣泛應用
作為機器學習的子集,深度學習在藥物探索領域得到了廣泛應用。例如,循環神經網路 (Recurrent Neural Network, RNN) 透過分子指紋技術發現了抗癌藥物。自動編碼器 (Autoencoder) 也被引入,透過直接對小型動物進行實驗來提供分子。
深度學習在藥物探索方面的廣泛應用,引起研究人員對開發更強大深度學習模型的濃厚興趣。然而,輸入數據 (如分子) 可以是任意大小和形狀的。目前,大多數機器學習和深度學習方法只能處理固定大小的輸入,即固定的向量、矩陣或張量。這一限制制約了機器學習方法在藥物發現方面的應用,特別是在毒性預測方面。
為了解決這個問題,一種名為圖神經網路 (Graph Neural Network, GNN) 的深度學習模型被提出。GNN 能夠以圖為輸入進行特徵提取,進行節點分類、連結預測、圖分類和圖生成等任務。例如,ChemRL 是一種有效的分子表徵學習方法,它成功地促進了分子性質的預測。(延伸閱讀│與輝達合作超級電腦後,華碩、國衛院如何靠AI改善醫療?)
實驗設計平台
為了對不同的毒性預測模型進行效能比較,我們採用了六個資料集:Tox 21、ClinTox、ToxCast、SIDER、HIV 和 BACE。
Tox 21 資料集是為測量化學化合物毒性而建立的公開資料集。它包含 7,950 種化合物以及 12 個毒性分類標籤。其中,我們隨機選取 6,242 個作為訓練集,683 個作為驗證集,784 個作為測試集。
ClinTox 是一個毒性預測資料集,由 1,451 種用於毒理學研究的化合物和 2 個毒性任務的標籤組成。在這些化合物中,我們隨機選擇了 1,182 個作為訓練集,148 個作為驗證集,148 個作為測試集。下圖顯示ClinTox 中的一些分子範例。
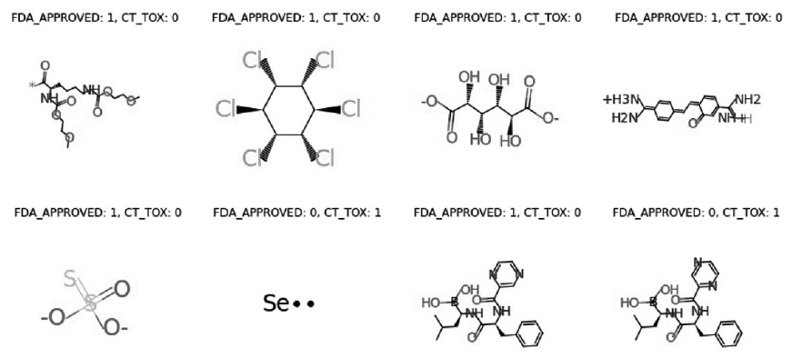
ToxCast 資料集是另一個公開的毒性預測資料集,提供了 8,575 個分子和 617 個分類任務。其中,我們隨機選取了 6,860 個樣本作為訓練集,857 個樣本作為驗證集,857 個樣本作為測試集。
SIDER 資料集是一個開源資料集,致力於匯總已上市藥物對人體的副作用。該資料集包含 1,427 種已獲批准的藥物和 27 項分類任務。我們將其中的 1141 種用於訓練,143 種用於驗證,其餘用於測試。
HIV 資料集是為實驗測量抑制 HIV 複製能力而建立的資料集。此資料集包括 41,127 個分子和 1 個分類任務。其中 32,901 個被用作訓練數據,4,113 個用於測試,另外 4,113 個用於測試集。
BACE 資料集包含一組人類 ß-分泌酶抑制劑的二元結合結果。此資料集包含 1,513 個分子和 1 個分類任務。訓練集的大小為 1,210 個,驗證為 151 個,測試為 152 個。
由於電腦處理所有結構和化學特徵的需求日益增長,多年來開發了大量化學分子表示法。化學結構通常用分子圖來表示:二維或三維表示法以及輸入字串:SMILES (簡化分子線性輸入系統)、Canonical SMILES、IUPAC 等。
根據不同的表示方法,這些分子會以不同的格式儲存。一旦分子以正確的格式表示,將三維表示法轉換為二維表示法或 SMILES 輸入字串就成為一個簡單的格式轉換問題。我們將 SMILES 字串作為分子編碼的輸入,並透過 Python 將其轉換為圖形表示。
下圖顯示從 Tox 21 中選取的分子 SMILES 字串及其對應的圖表示 (即鄰接矩陣)。最後也展示了所選分子的可視化效果。透過將分子表示為圖形,我們可以包含分子的拓撲資訊、節點及其鄰接關係,甚至連通性屬性。
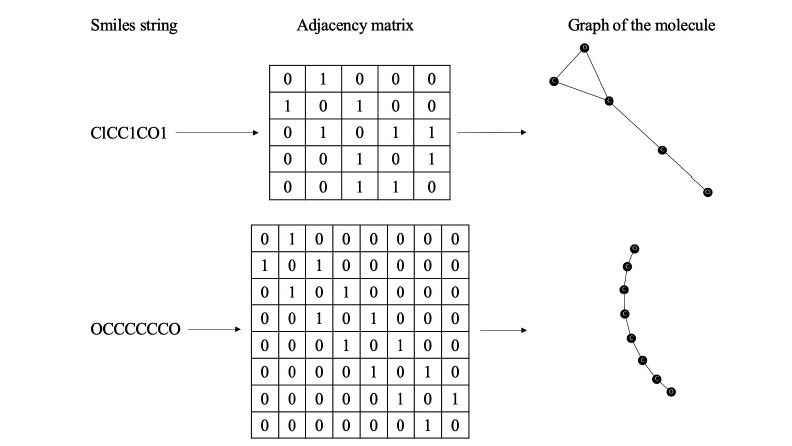
透過這種方法,我們可以盡可能保留分子的資訊,這無疑有利於分子的性質預測。
在實驗中,我們為隨機森林創建了 500 棵決策樹,並採用 Gini 不純度來分割節點以形成決策樹。對於 MLP,我們建立了一個三層感知器,並在輸出層末端使用 Softmax 分類器執行了 300 個訓練週期(Epoch)。
我們的所有實驗都是透過 Pytorch 使用四個NVIDIA Titan XP GPU 實現的。此外,圖卷積和 GIN 由 TorchDrug 實現,它在 Pytorch 中提供了藥物探索模型。
結論
本文將機器學習方法應用於藥物特性預測任務,特別是毒性預測。我們展示了深度學習方法,尤其是圖神經網路 (GNN),在學習化學分子的圖表示方面擁有卓越能力。
GNN 能夠將分子視為任意大小和形狀的圖,並透過定位節點和邊來聚合圖嵌入,從而比其他機器學習方法取得更好的效果。
從不同表現指標的實驗結果來看,我們觀察到,分類準確率在其他應用領域中常用,但可能並不適合藥物性質預測。因為我們預測藥物性質的主要目標是明確區分陽性樣本和陰性樣本。因此,使用 ROC AUC 分數是衡量藥物特性預測模型效能的一個更好的方法。
ROC AUC 分數被用來衡量毒性預測的效能,計算的是 ROC 曲線下的面積,該曲線顯示每個閾值下的真陽性率 (TPR) 和假陽性率 (FPR)之間的權衡。
總結來說,我們的研究表示,深度學習方法在藥物毒性預測中的應用具有顯著優勢。GNN 尤其在處理圖結構數據方面表現出色,能夠在多個資料集上取得優異的預測性能。
透過進一步調整超參數和改進模型結構,我們相信 GNN 在藥物性質預測中的應用潛力將得到進一步發揮。這一成果為未來藥物開發和毒性評估提供了新的方向和方法。
(延伸閱讀│台灣將有另一座「護國神山」?AI聯手醫療雙強升級)